
REVISIÓN
Sistema renina-angiotensina (SRA) en las patologías cardiovasculares: papel sobre la hipertensión arterial
Renin-angiotensin system (RAS) in cardiovascular pathologies: role on arterial hypertension
Norma Ciau-Solís, David Betancur-Ancona
Facultad de Ingeniería Química, Universidad Autónoma de Yucatán. Periférico Norte Km. 33.5, Tablaje Catastral 13615, Colonia Chuburná de Hidalgo Inn, 97203 Mérida, Yucatán, México
* Autor para correspondencia.
![]()
|
|
Attribution-NonCommercial-ShareAlike 4.0 International License La revista no cobra tasas por el envío de trabajos, |
|
Resumen
Introducción. En las patologías cardiovasculares como insuficiencia cardiaca, hipertensión arterial, vasculopatías, enfermedades coronarias, etc; el sistema renina-angiotensina (SRA) tiene participación clave, cuyas acciones principales incluyen la regulación de la presión arterial, el tono vascular, la volemia y facilitar la transmisión simpática.
Objetivo. En este ensayo se discuten estos aspectos del SRA como resultado de una secuencia de transformaciones de distintas proteínas, comenzando por la acción de la renina que transforma el angiotensinógeno en angiotensina-I y posteriormente, este se convierte en angiotensina II por acción de la enzima convertidora de angiotensina I (ECA I).
Conclusiones. Cuando este sistema se ve alterado culmina en hipertensión arterial, la cual se puede controlar mediante tratamientos farmacológicos empleando inhibidores químicos cuyos mecanismos de acción se basan en impedir que estas enzimas se unan a su sustrato y así se mantenga un equilibrio homeostático en la presión arterial.
Palabras clave
Renina; Angiotensina; hipertensión; inhibidores
Abstract
Introduction. In cardiovascular pathologies such as heart failure, arterial hypertension, vasculopathies, coronary diseases, etc; the renin-angiotensin system (RAS) has an essential role. The main actions include the regulation of blood pressure, vascular tone, volemia and facilitating sympathetic transmission.
Objective. This manuscript summarizes these aspects of the SRA and discussed the sequence of transformations of different proteins, beginning with the action of renin that transforms the angiotensinogen into angiotensin-I and later, it is converted into angiotensin II by the action of the Angiotensin-I converting enzyme (ACE-I).
Conclusions. When this system is altered culminates in hypertension, which can be controlled by pharmacological treatments using chemical inhibitors whose action mechanisms are based on preventing these enzymes from binding to their substrate and thus maintain a homeostatic balance in the pressure arterial.
Keywords
Renin; Angiotensin; hypertension; inhibitors
Introducción
En las patologías cardiovasculares, insuficiencia cardiaca, HTA, vasculopatías, enfermedades coronarias, el SRA tiene participación esencial de manera particular en la regulación de la PA, el tono vascular, la volemia y para facilitar la transmisión simpática. El SRA participa en la remodelación ventricular del hipertenso o del infartado, así como en la remodelación vascular que consiste en la capacidad de una arteria en adaptar su tamaño estructural ante estímulos crónicos por crecimiento o reducción del tamaño externo para mantener un lumen funcional (1).
El SRA clásico o plasmático (Figura 1) tiene un importante papel en la regulación de la presión arterial (PA) por medio de la liberación de Ang II y el equilibrio hidrosalino, a través de la liberación de aldosterona. Normalmente, el SRA actuaría como un mecanismo de defensa que se activaría en respuesta a una hipotensión hipovolémica. Cuando la PA disminuye como resultado de la restricción de sodio o hipovolemia, las células yuxtaglomerulares del riñón sintetizan una enzima, la renina, que se libera a la sangre circulante (2).
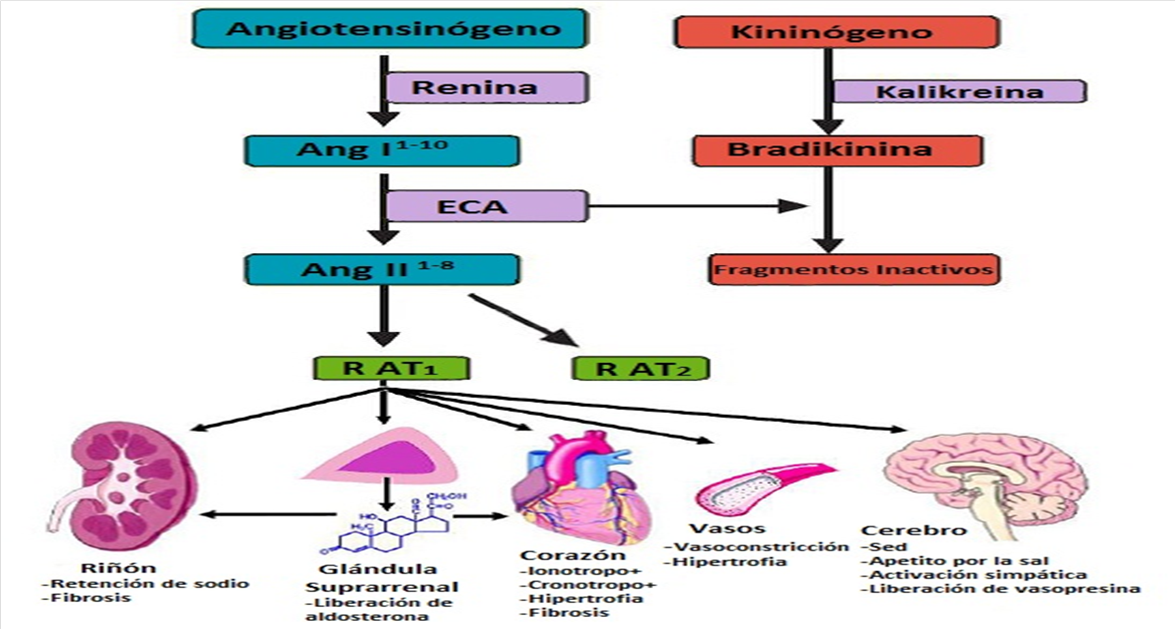
Figura 1. SRA clásico. Angiotensina (Ang); Enzima convertidora de angiotensina (ECA); Receptor de la Angiotensina 1 (R AT1) y Receptor de la Angiotensina 2 (R AT2). (Ponce & Ponce, 2012).
La renina rompe el enlace existente entre Leu-10 y Val-11 del angiotensinógeno (AGT), glucoproteína β2 plasmática de 453 aminoácidos (55-61 kDa) sintetizada en el hígado, lo que produce un decapéptido inactivo, la Ang I, la cual recibe la acción de la ECA, secretada por las células endoteliales de los pulmones, fundamentalmente, y de los riñones, e hidroliza el dipéptido terminal His-9–Leu-10 de la Ang I y la convierte en un octapéptido activo, la Ang II. Esta estimula los receptores AT1 y AT2, y produce una respuesta vasoconstrictora (que incrementa las resistencias vasculares periféricas y la PA), aumenta la actividad del sistema simpático, estimula la liberación de vasopresina, incrementa el cronotropismo cardíaco, y favorece la aparición de cambios en las funciones glomerular y tubular del riñón, con un aumento en la liberación de aldosterona por la corteza suprarrenal que es la que produce retención renal de sodio y agua, con lo que aumenta aún más la PA(3).
Estas acciones permiten restaurar la volemia y la PA. A su vez, la propia Ang II estimula los receptores AT1 en las células yuxtaglomerulares y produce una inhibición de la síntesis y la liberación de renina a este nivel, con lo que se regula la activación del SRA.
Enzimas que participan en el SRA
Renina
La renina es una proteasa de aspartilo sintetizada en la forma de una proenzima inactiva, la prorrenina. Gran parte de la renina en la circulación es sintetizada en las arteriolas renales aferentes. La prorrenina puede ser secretada en forma directa a la circulación o ser activada dentro de las células secretoras y liberada en la forma de renina activa (4).
El plasma humano contiene de dos a cinco veces más prorrenina que renina, pero no hay datos de que la primera contribuya a la actividad fisiológica de tal sistema. Se han identificado tres estímulos primarios de la secreción de renina: 1) menor transporte de cloruro de sodio en la región distal de la rama ascendente gruesa del asa de Henle, que está en relación directa con la arteriola aferente correspondiente (mácula densa); 2) disminución de la presión o el estiramiento dentro de la arteriola renal aferente (mecanismo baroreceptor), y 3) estimulación de tipo simpático de las células reninógenas a través de receptores adrenérgicos β1. Por lo contrario, el aumento del transporte de cloruro de sodio en la porción ascendente gruesa del asa de Henle inhibe la secreción de renina, por un mayor estiramiento dentro de la arteriola aferente renal y por antagonismo de los receptores β1. También, la angiotensina II puede inhibir directamente la secreción de renina, a causa de la acción de sus receptores de tipo 1 en las células yuxtaglomerulares, sin embargo, la secreción de renina aumenta en reacción al antagonismo farmacológico con ECA-I o antagonistas de receptores de angiotensina II (5).
La renina activa es secretada del aparato yuxtaglomerular del riñón, una vez liberada a la circulación, desdobla un sustrato, el angiotensinógeno (glucoproteína de 452 aminoácidos), para formar un decapéptido inactivo, la angiotensina I, mediante la ECA-I, que se encuentra en la circulación pulmonar (aunque no en forma exclusiva) y que convierte la angiotensina I en el octapéptido activo, angiotensina II (6, 7).
Fármacos inhibidores de renina
Los inhibidores de la renina se consideran actualmente como un nuevo enfoque en el tratamiento de la HTA. La renina ha sido ampliamente reconocida como el sitio preferido para el bloqueo del SRA porque participa en el primer paso para la conversión del angiotensinógeno en angiotensina I. El descubrimiento del receptor de prorrenina y renina constituye una razón adicional para centrar la atención en la inhibición de la renina. Cuando se une la renina a su receptor, su actividad enzimática se amplifica ejerciendo efectos fisiológicos que son enteramente independientes de la producción de Angiotensina II. Además, la pro-renina, que es simplemente un precursor inactivo de la renina, se convierte en una droga biológicamente activa cuando está unida a este receptor (8). Tanto la renina como la prorrenina pueden activar señales intracelulares produciendo estímulos profibróticos y de vasoconstricción, independientes de la Angiotensina II. La aprobación por la FDA del Aliskiren en Marzo del 2007 constituye un nuevo enfoque en la supresión o inhibición del SRA para el tratamiento de la presión arterial (9, 10).
El Aliskiren, es un inhibidor competitivo no peptídico de la renina por vía oral, que se une al sitio activo de la molécula de renina, bloqueando la fragmentación del angiotensinógeno y previniendo la formación de angiotensina I. Numerosos estudios clínicos han demostrado al menos equivalencia o una mayor disminución de la presión arterial comparados con las drogas existentes, pero con menos efectos colaterales. Aliskiren, posee efectos sinérgicos cuando se combina con diuréticos tiazídicos, inhibidores de ECA, bloqueadores de los receptores de angiotensina y bloqueadores de los canales de calcio, ambos en términos de eficacia y tolerabilidad (11, 12).
Química y mecanismo de acción de los inhibidores de renina
El aliskiren es una sal de hemifumarato 2-carbamil-2-metilpropil-5 amino-4 hidroxi-2-7-disopropil-8-4metoxipropoxifeniloctamida, y es un potente inhibidor de renina no peptídico con concentración inhibitoria de 0.6 nm/l para la inhibición in vitro de la renina humana. Cuando se produce la unión del aliskiren con el sitio activo de la renina (S1/S3), se bloquea la actividad de Asp32 y Asp215 de residuos de aspartato previniendo así la conversión del angiotensinógeno en angiotensina I (Figura 2). El aliskiren es una molécula hidrofílica, con una alta solubilidad al agua, que facilita su biodisponibilidad oral (11). Existen 2 términos que deben mencionarse en esta revisión. Uno de ellos es la Actividad de la Renina Plasmática (ARP), reportada en ng/ml/hora y es la frecuencia con que la angiotensina I es producida después de la adición de angiotensinógeno en el plasma y el otro término es la Concentración de Renina Plasmática (CRP), reportada en pg/ml y mide la cantidad de renina y prorenina juntos en el plasma. Con la excepción de los betabloqueadores, todos los inhibidores del SRA disponibles hasta la fecha incrementan la CRP por disminución de la AG II. Sin embargo, los inhibidores de renina disminuyen la ARP, a diferencia del resto de inhibidores actuales del SRA. Se ha reconocido que el incremento de la ARP está relacionada con un incremento de eventos cardiovasculares y cerebrovasculares en estudios epidemiológicos realizados los últimos 20 años (9, 10).
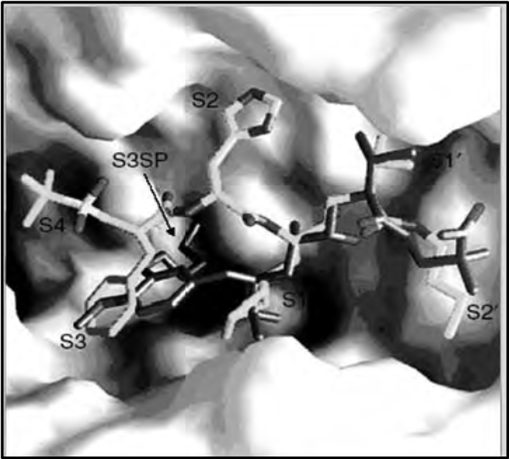
Figura 2. Estructura del Aliskiren. Cristalografía general del Aliskiren en el complejo formado en la renina humana. Unión del Aliskiren con el sitio activo de la renina humana en los sitios de especificidad enzimática (Bustamente, 2008).
Desventajas de los inhibidores de la Renina
La administración de aliskiren en monoterapia al parecer tiene la misma eficacia de los IECA-I para disminuir la presión arterial, pero no es mejor que éstos. Es posible alcanzar disminuciones mayores de la presión cuando se combina el aliskiren con un diurético tiazídico, un IECA-I, un ARA-II o antagonistas del calcio. En la actualidad, se considera que el aliskiren, el cual se comercializa como Tekturna® y/o Rasilez® en gran parte del mundo, no es un antihipertensivo de primera línea, tampoco se han realizado ensayos clínicos sobre sus efectos en complicaciones cardiovasculares o renales mórbidas o mortales en la HTA y está contraindicado en pacientes diabéticos (9).
Ezima Convertidora de Angiotensina I (ECA-I)
La ECA-I es una ectoenzimametaloproteinasa de zinc dipeptidicarboxipeptidasa (quininasa II, EC 3.4.15.1) que escinde el dipéptido histidil-leucina de la terminación carboxilo del decapéptido DRVYIHPFHL de la Angiotensina-I, mediante el SRA generando el octapéptido vasoconstrictor angiotensina-II, y por otro lado hidroliza el péptido vasodilatador bradikinina, en su función de kininasa II, mediante el sistema kalikreina-kinina (6,13) . Está ubicada en la membrana de las células endoteliales (CE) parenquimatosas y también en las inflamatorias. En el ser humano, la ECA-I se presenta en dos isoformas; la primera ha sido denominada ECA somática (sECA) y se localiza en las regiones antes descritas. La otra isoforma, denominada ECA germinativa o testicular (tECA), es una enzima más pequeña, se localiza en células germinativas en el testículo y su acción está relacionada con la maduración del esperma en varones. Existe además otra forma, denominada soluble, que se ha aislado mediante la acción de una secretasa y se ha encontrado en suero sanguíneo y otros fluídos corporales; sin embargo, aún no se esclarece su participación fisiológica en el ser humano. La sECA se caracteriza por tener dos dominios homólogos que se denominan de acuerdo a su posición en la cadena: N-terminal o Dominio-N y C-terminal o Dominio- C, conteniendo ambos zinc en el sitio de unión y un centro activo. La tECA solamente tiene el Dominio-C. Ambas isoformas de la ECA se hallan entre la estructura de la membrana plasmática, en la cual la parte del Dominio-C se halla en la extensión intracelular, en tanto que la mayor parte de la cadena de la ECA permanece fuera de la célula; por esta razón se les ha denominado ectoenzimas cuya misión es hidrolizar péptidos circulantes a través de la parte exterior de las células. Una primera representación esquemática de los dominios de las 3 formas de la ECA se muestra en la Figura 3 (14).
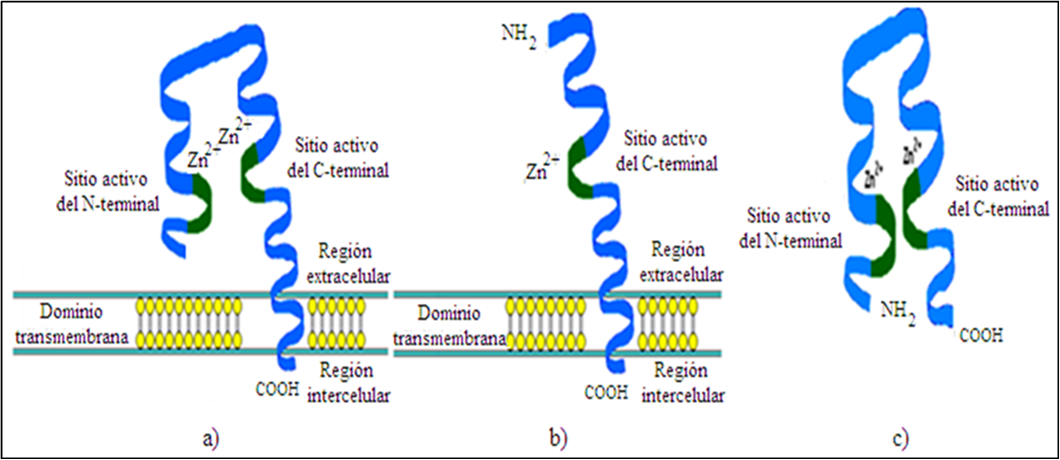
Figura 3. Estructura y conformación de la ECA-I. Encontrada en células somáticas (a), testicular (b) y plasma (c), mostrando los sitios activos catalíticos, dependencia del zinc y las regiones N y C terminal.
Sitio activo y dominios de la ECA-I
Cada dominio de la ECA tiene un motivo enlazador de zinc típico, denominado His-Glu-X-X-His en el sitio activo, similar al hallado en muchas enzimas zinc peptidasas. Los dos residuos de histidina proveen dos de los tres ligandos del zinc y el grupo carboxilo del residuo de glutamato es la base donante de electrones en la reacción catalítica. La sECA humana tiene 2 sitios activos que se encuentran entre 2 dominios homólogos que tienen varias características que diferencian los 2 sitios catalíticos, por ejemplo, la activación con ion cloro (15).
En la secuenciación de ambos dominios catalíticos N y C, se hallan 2 residuos de prolina en promedio, por cada espacio de 23 residuos. En las regiones repetidas se contaron 10 residuos de cisteína. En la tECA, hay tres puentes disulfuro adyacentes (aabbcc) con una cisteína libre localizadas entre el Dominio-C y la parte media de todos los puentes disulfuro, confirmadas con cristalografía de rayos X. La secuencia de aminoácidos de ambos dominios difieren en la longitud de sus cadenas por 7 residuos; la secuencia es homóloga entre ellos, en un 55 %, aproximadamente. En el canal donde se ancla el sustrato, las diferencias están marcadas en las regiones de proteína. Todas las estructuras del Dominio-N, al igual que el Dominio-C, son estructuras secundarias. El Dominio-N tiene 27 estructuras de hélice (Figura 3), de las cuales 18 son α-hélice, 5 son hélices cortas de la forma α-310 y 4 son hélices de longitud intermedia; en este Dominio, solamente se han hallado 6 estructuras β-plegada (14).
Mecanismo de acción de la ECA-I y afinidad por el sustrato
En 1987, Büning presentó un modelo hipotético del sitio activo de la ECA-I y la interacción de un péptido como sustrato. En este modelo (Figura 4), desarrollado a partir de experimentos de modificaciones químicas a la enzima, se propuso que los residuos de arginina, ácido glutámico y tirosina fueron los componentes catalíticos del sitio activo. El sustrato interactúa a través del grupo carbonilo con el átomo esencial de zinc, facilitando el ataque nucleofílico del grupo carboxilo del ácido glutámico. La subsecuente adición de moléculas de agua completa el anclaje y ligación del péptido como sustrato. El residuo de tirosina dona los protones al grupo NH del péptido, mientras que la especificidad es determinada por la interacción del grupo carboxilo terminal con el residuo de arginina (14).
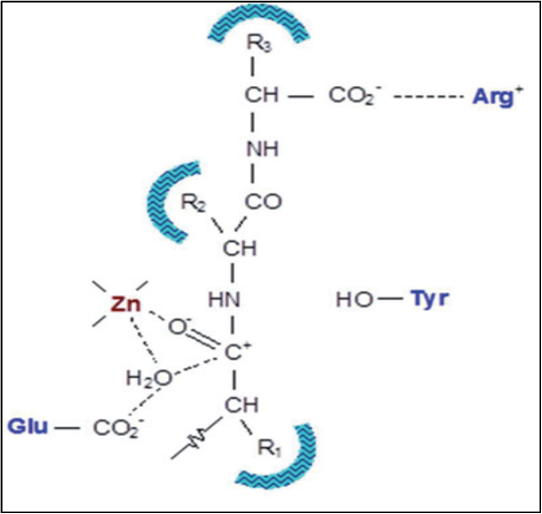
Figura 4. Modelo hipotético del sitio activo de la ECA-I y su interacción con un péptido como sustrato (Domínguez et al., 2012).
Investigaciones posteriores revelaron que la ECA-I tiene propiedades de endo y exopeptidasa, la actividad inusual de exopeptidasa se observó mediante el anclaje e hidrólisis del segundo residuo de aminoácido en la región terminal del grupo carboxilo en una variedad de péptidos como sustrato, como la bradikinina, angiotensina-I, HHL (Hipuril-L-Histidil-L-Leucina) y Z-FHL. Se ha demostrado que un tripéptido con la región amino terminal acilado es el mejor sustrato para la ECA-I, como el caso del tripéptido Z-FHL, con terminación análoga a la angiotensina-I. Así mismo, la presencia de un residuo de aminoácido aromático como la Fen en la antepenúltima posición confiere al péptido sustrato un gran valor de afinidad con la enzima. La mayor afinidad de la enzima ECA-I se presenta con el sustrato bradikinina(9). Actualmente se sabe que la actividad primaria de la ECA es anclarse en oligopéptidos que presenten un grupo carboxilo libre con amplia especificidad. Sustratos conteniendo Pro en la posición P1’ (nomenclatura de Schechter y Berger, 1967) y Asp o Glu en la posición P2’ son resistentes al ataque de la ECA. Los Dominios C y N terminal exhiben actividades catalíticas similares para hidrolizar a la angiotensina-I, bradikinina y la sustancia P. Los estudios realizados in vitro, han demostrado que la inhibición completa al ataque de la ECA en la angiotensina I y la bradikinina requiere el bloqueo de ambos Dominios; en contraste, los estudios in vivo con ratas de laboratorio, han revelado que la inhibición de cualquiera de los Dominios N y C no permite la hidrólisis de la angiotensina-I a angiotensina-II, en tanto que para evitar la hidrólisis de la bradikinina se necesita la inhibición de ambos Dominios a la vez en forma simultánea (14).
Fármacos inhibidores de la Enzima Convertidora de Angiotensina I (IECA-I)
El 1965, investigaciones científicas demostraron que el extracto etanólico no tóxico, aislado a partir del veneno de una serpiente brasileña, Bothrops jararaca, potencializaba la contracción del músculo liso inducido por la bradikinina; debido a esta propiedad, a este extracto etanólico se le denominó “factor de potencialización de bradikinina” (BPF), concluyendo después de estudiar sus propiedades, que se trataba de un péptido o mezcla de ellos. Después de varias investigaciones, se pudo conocer que son oligopéptidos con 5 o 13 aminoácidos constituyentes; así se dio inicio a la construcción de drogas para la inhibición de la ECA-I, al tratar de diseñarlas con la misma secuencia de aminoácidos .
El estudio de la estructura de los fármacos y la de los péptidos del veneno de la serpiente, indica que la región C-terminal contribuye significativamente para enlazarse en el sitio activo de la ECA-I. Se ha visto que la secuencia óptima de la región C-terminal para enlazarse en el sitio activo es WAP, sin embargo el triptófano usualmente se reemplaza por un aminoácido más estable como la fenilalanina. Estos aminoácidos han demostrado tener las interacciones óptimas con los subsitios hipotéticos S1, S1’ y S2’ del sitio activo de la ECA-I. Se han estudiado fármacos y drogas que han mostrado resistencia a la digestión gastrointestinal después de la ingesta oral y han tenido una gran afinidad por el sitio activo de la ECA-I, y de los cuales se han producido dipéptidos, siendo el captopril el mejor representante de este grupo, considerado un análogo del dipéptido Ala-Pro de la región C-terminal del péptido aislado del veneno de la serpiente brasileña BPP5a, en la cual la función amina de la alanina es reemplazada por el grupo sulfhidrilo, el cual interactúa con mayor fuerza con el ión zinc catalítico de la ECA-I. Adicionalmente, el grupo carboxilo terminal interactúa con la carga positiva de la arginina que se localiza en el sitio activo de la ECA-I, en tanto que el grupo metilo lo hace con el subsitio S1’ (Figura 5) (3).
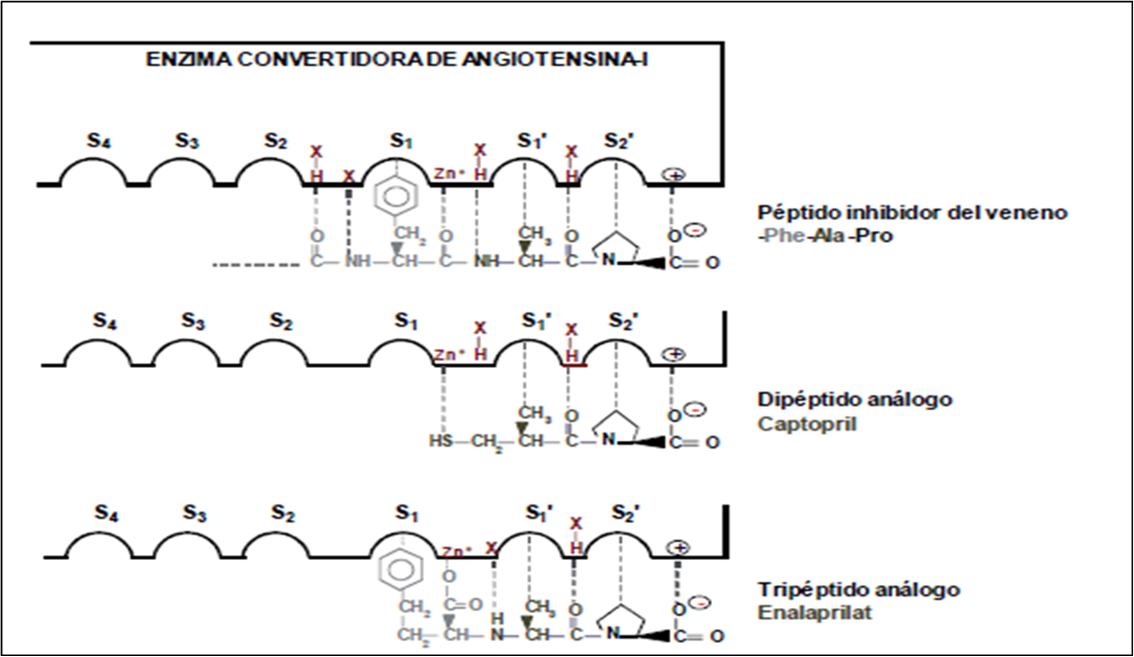
Figura 5. Enlaces hipotéticos de los inhibidores competitivos con el sitio activo de la ECA-I. Los subsitios denotados por S1, S1’, S2’ interactúan con el último, penúltimo y antepenúltimo residuo de aminoácido de la cadena constituyente del inhibidor en cuestión. El residuo X-H es un donador de hidrógeno, en tanto que X es un aceptor de hidrógeno.
Muchos de los fármacos inhibidores de la ECA-I son análogos a la región terminal del tripéptidos Fen BPP5a, denominados inhibidores tripeptídicos análogos. El metabolito enalaprilat es la forma ácida activa liberada a partir del fármaco enalapril, el cual se administra como una prodroga éster y el metabolito se libera después de la digestión intestinal. El enalaprilat se une con el sitio activo de la ECA-I empleando las demás interacciones del residuo del BPP5a en la región C-terminal, reemplazando la débil interacción de su región amida carbonilo con el ión zinc de la enzima, por la interacción fuerte del grupo carboxilo del enalaprilat. Esta función del carboxilo también evita que el enalaprilat sea hidrolizado por la propia ECA-I. Bajo condiciones fisiológicas in vivo (empleando ratas normotensas), el enalaprilat es más activo que el captopril (Tabla 1) (14).
Tabla 1. Potencia de los fármacos más empleados en la inhibición de la ECA-I.
|
Fármaco inhibidor |
IC50 en nM |
|
Captopril Enalapril Enalaprilat Lisinopril Ramipril |
9.7 4645.0 2.8 1.4 616.0 |
|
Ramiprilat |
0.7 |
La farmacoterapia es recomendable en personas con presiones arteriales ≥140/90 mmHg (16). Estos fármacos han sido considerados como inhibidores competitivos, reversibles y de rápida acción para enlazarse con la ECA-I y provocar inhibición. El grado de beneficio obtenido de tales fármacos depende de la magnitud de la disminución de la presión arterial. La disminución de 10 a 12 mmHg de la PAS y de 5 a 6 mmHg en la PAD confiere las disminuciones relativas de riesgo de 35 a 40% para el caso de apoplejía y 12 a 16% para cardiopatía congestiva en término de cinco años de haber comenzado el tratamiento (4).
Enzima convertidora de angiotensina II
En el año 2000, se identificó una nueva enzima homóloga de la ECA-I, a la cual se denominó Enzima convertidora de angiotensina II (ECA-II). Esta enzima es homóloga en un 42% con la ECA-I, pero con actividades bioquímicas diferentes. La ECA-II al hidrolizar a la angiotensina I genera angiotensina (1-9 aminoácidos), la cual sirve como una vía indirecta para generar angiotensina II; sin embargo, la actividad catalítica de ECA-II es 400 veces mayor sobre la angiotensina II que sobre la angiotensina I, y conlleva a la formación de angiotensina (1-7 aminoácidos) con propiedades vasodilatadoras. De esta manera el SRA puede ser visto como un sistema endocrino dual en el que las acciones vasoconstrictoras y vasodilatadoras son reguladas por un balance entre la ECA–I y la ECA-II, lo cual hace fácilmente entendible el efecto benéfico que tienen los inhibidores de la enzima convertidora de angiotensina I (ECA-I) en el perfil de pacientes cardiometabólicos (6).
Conclusiones
Si bien es conocido que el SRA no es el único mecanismo de regulación de la presión arterial, sí es importante recalcar que juega un papel primordial en esta. El entendimiento de los mecanismos de acción de las enzimas responsables es fundamental para promover el adecuado control de las patologías cardiovasculares, específicamente la hipertensión arterial, pudienso ser el el empleo de sustancias o fármacos que actúen como inhibidores de Renina o como inhibidores de la ECA-I, mejor conocidos como IECA.
Referencias
1. De la Serna, F. Novedades en el sistema renina-angiotensina. Insuficiencia Cardiaca, 2014; 9(1), 16-24.
2. Nawaz KAA, David SM, Murugesh E, Thandeeswaran M, GopikrishnanKiran K, Mahendran R. et al. Identification and in silico characterization of a novel peptide inhibitor of angiotensin converting enzyme from pigeon pea (Cajanus cajan). Phytomedicine, 2017, 9:13.
3. Ponce Y, Ponce A. El sistema Renina Angiotensina desde la circulaicón hasta la célula: implicaciones más allá de la hipertensión. CorSalud, 2012; 4(4):287-293.
4. Kotchen, T. Vasculopatía hipertensiva. In D. Longo, A. Fauci, D. Kasper, S. Hauser, J. Jameson & J. Loscalzo Eds., HARRISON Principios de medicina interna. New York, N.Y. USA: McGraw-Hill. 2012.
5. Ames MK, Atkins CE, Pitt B. The renin-angiotensin-aldosterone system and its suppression. J Vet Intern Med. 2019; 33:363–382.
6. Lima M, Nuccio J, Villalobos M, Balladares N. Sistema renina angiotensina y riesgo cardiometabólico. Rev. Venezolana Endocrin. Metab, 2010; 8(1):3-10.
7. Macaulay A, Nnonyelum T. Angiotensin converting enzyme inhibitors. In A. De Brue Ed., Angiotensin converting enzyme inhibitors. New York, NY, USA.: Nova Science Publishers, Inc. 2009.
8. Bader M. Tissue renin-angiotensin-aldosterone systems: targets for pharmacological therapy. Annu Rev Pharmacol Toxicol. 2010; 50(1): 439-465
9. Bustamante, G. Inhibidores de renina. Rev. Peruana Cardiol, 2008; 34(2): 129.
10. Aluko RE. Food protein-derived renin-inhibitory peptides: in vitro and in vivo properties. J Food Biochem. 2019; 43:12648.
11. Chen Y, Meng L, Shao H, Yu F. Aliskiren vs. other antihypertensive drugs in the treatment of hypertension: A meta-analysis. Hypertension Research, 2013; 36:252–261
12. Morales F, Estañ, L. Aliskiren: el primer inhibidor directo de la renina introducido en terapéutica. Rev Esp Cardiol Supl, 2009; 9:41A-48A
13. Bleakley S, Hayes M, O’Shea N, Gallagher E, Lafarga, T. Predicted release and analysis of novel ACE-I, renin, and DPP-IV inhibitory peptides from common oat (Avena sativa) protein hydrolysates using in silico analysis. Foods, 2017; 6:108
14. Domínguez M, Betancur D, Chel L. Caracterización de la ECA-I e inhibición con péptidos alimentarios. Alemania: Editorial Académica Española. 2012.
15. Guang C, Phillips RD, Jiang B, Milani F. Three key proteases-Angiotensin-I-converting enzyme (ACE), ACE2 and Renin- within and beyond the renin-angiotensin system. Arch. Cardiovasc. Dis. 2012; 105:373–385.
16. Yang Y, Tao G, Liu P, Liu J. Peptide with angiotensin I-converting enzyme inhibitory activity from hydrolyzed corn gluten meal. J. Agric. Food Chem, 2007; 55(19): 7891-7895